(jee-no-fee)
Genotype-to-Phenotype Phage-Host Interaction Prediction
GenoPHI is a Python package for machine learning-based prediction of genotype-phenotype relationships using whole-genome sequence data. Originally designed for phage-host interaction prediction, GenoPHI supports both binary interaction prediction and regression tasks for any microbial phenotype. The package implements protein family-based and k-mer-based approaches to extract genomic features from amino acid sequences and predict phenotypes using CatBoost gradient boosting models.
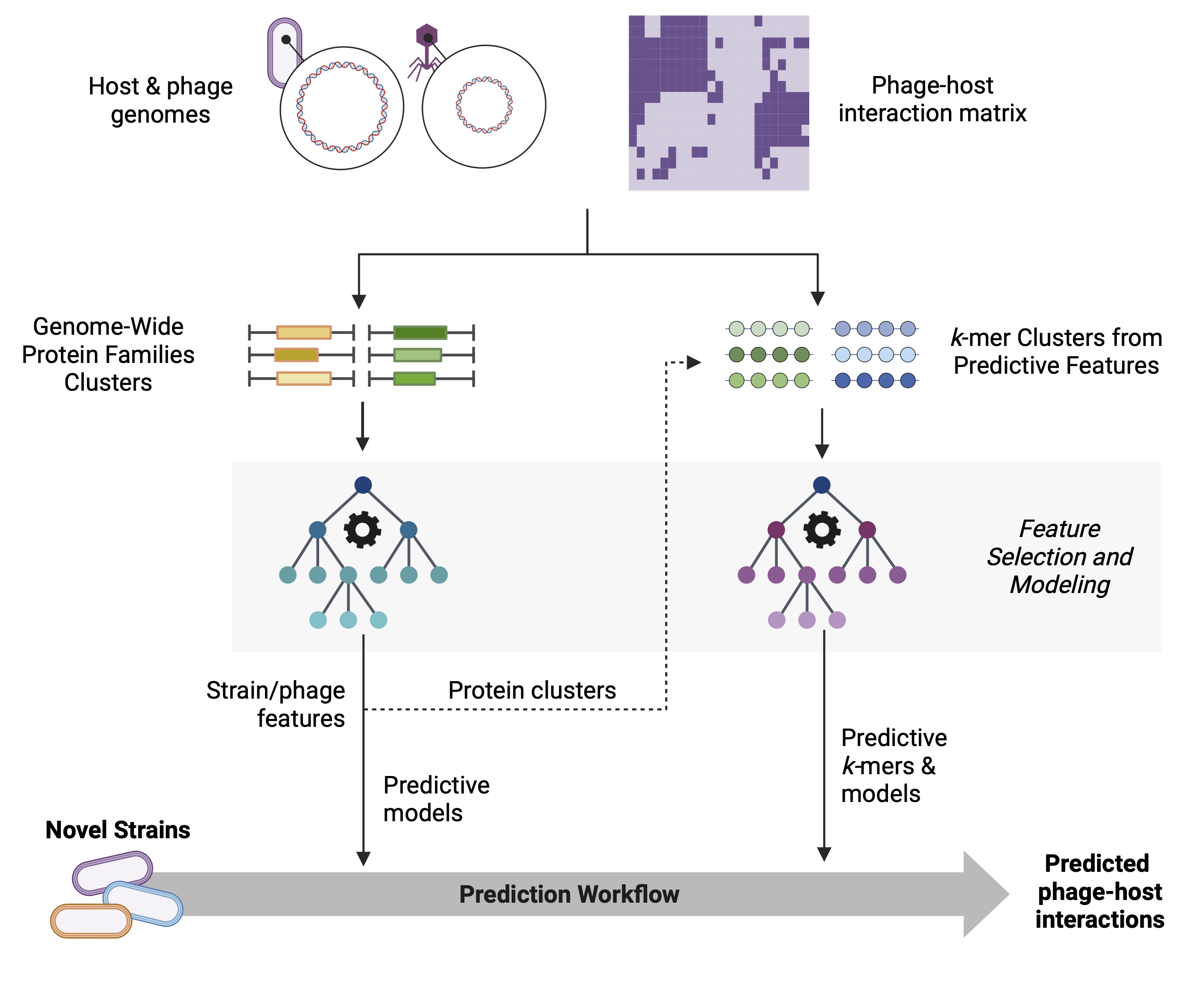 Figure 1: GenoPHI workflow schematic showing the main analysis pipelines: Protein family-based workflow, K-mer-based workflow, and Predictive protein k-mer workflow. Each pathway includes feature extraction, selection, model training, and prediction steps.
Figure 1: GenoPHI workflow schematic showing the main analysis pipelines: Protein family-based workflow, K-mer-based workflow, and Predictive protein k-mer workflow. Each pathway includes feature extraction, selection, model training, and prediction steps.
- Features
- Installation
- Quick Start
- Usage
- Input Data Formats
- Feature Selection Methods
- Performance Metrics
- Output Directory Structure
- Python API
- Advanced Usage
- Troubleshooting
- Testing
- Best Practices
- Citation
- Contributing
- License
- Support
- Acknowledgments
- MMSeqs2 Clustering: Cluster protein sequences into protein families based on sequence similarity
- Feature Table Generation: Create presence-absence matrices of protein families across genomes and consolidate into predictive features based on co-occurence across genomes
- Feature Selection: Identify predictive protein families (multiple available methods: RFE, SHAP, SHAP-RFE, ANOVA, Chi-squared, Lasso)
- Model Training: Train CatBoost models with hyperparameter optimization
- Phenotype Prediction: Predict interactions, resistance, or other phenotypes for new genomes
- Feature Annotation: Identify predictive protein sequences from predictive features
- K-mer Feature Extraction: Generate k-mer features from protein sequences with or without gene family context
- Predictive K-mer Workflow: Extract k-mers specifically from predictive protein families identified in protein family analysis
- Feature Selection & Modeling: Apply same robust feature selection and modeling pipelines
- Flexible K-mer Lengths: Support for single k-mer length or ranges (e.g., 3-6)
- Phage-Host Interaction Prediction: Binary prediction of infection outcomes between phages and bacterial strains
- Single-Strain Phenotype Prediction: Predict strain-level phenotypes (e.g., antibiotic resistance, growth rate) without requiring phage data
- Regression Tasks: Predict continuous phenotypes (e.g., infection efficiency, metabolic rates)
- General Feature-Based Modeling: Use any feature table with a phenotype column for custom applications
- Dynamic Feature Weighting: Account for feature frequency distributions to handle imbalanced features
- Clustering-Based Selection: Use HDBSCAN or hierarchical clustering for intelligent feature grouping
- Multiple Feature Selection Methods: RFE, SHAP-RFE, SelectKBest, Chi-squared, Lasso, SHAP
- Comprehensive Performance Metrics: AUC-ROC, Precision-Recall, MCC, F1-score, Accuracy
- SHAP Interpretability: Feature importance analysis and visualization for model explainability
- Bootstrapping Support: Robust model evaluation with multiple train-test splits
Minimum Requirements:
- Python 3.8 or higher
- 8 GB RAM
- 4 CPU cores
- 10 GB free disk space
Recommended for Large Datasets:
- Python 3.10+
- 32+ GB RAM
- 8+ CPU cores
- 50+ GB free disk space (depending on dataset size)
Tested Operating Systems:
- Linux (Ubuntu 20.04+, CentOS 7+)
- macOS (Sonoma 14+, Apple Silicon)
Create and activate a conda environment:
conda create -n genophi python=3.10
conda activate genophiFrom PyPI (Recommended):
pip install genophiFrom GitHub (Development):
git clone https://github.com/Noonanav/GenoPHI.git
cd GenoPHI
pip install -e .For development with optional dependencies:
pip install -e ".[dev]"External Dependency: GenoPHI requires MMseqs2 for protein sequence clustering and assignment.
Install via conda/mamba:
conda install -c bioconda mmseqs2
# or
mamba install -c bioconda mmseqs2For other installation methods, see the MMSeqs2 Wiki.
Test that GenoPHI is properly installed:
# Check GenoPHI version
genophi --version
# Verify MMseqs2 is accessible
mmseqs version
# Run basic help command
genophi --helpFull installation (conda environment + GenoPHI + MMseqs2) takes approximately 2-3 minutes on a standard desktop computer (tested on a MacBook Pro M2, 16 GB RAM, macOS Sonoma 14.3).
A small test dataset is included in the repository for demonstrating the software. To run the demo:
git clone https://github.com/Noonanav/GenoPHI.git
cd GenoPHI
genophi protein-family-workflow \
--input_path_strain data/test_data/strain_AAs/ \
--input_path_phage data/test_data/phage_AAs/ \
--phenotype_matrix data/test_data/ecoli_test_interaction_matrix.csv \
--output_dir demo_output/ \
--threads 4 \
--num_features 50 \
--num_runs_fs 5 \
--num_runs_modeling 10 \
--method rfe \
--filter_type strainTest dataset: 25 E. coli strains and 25 phages with a binary interaction matrix.
Expected output: A demo_output/ directory containing MMseqs2 clustering results, feature selection outputs, trained models, performance metrics, and a workflow summary report. See Output Directory Structure for details.
Expected run time: ~25 minutes on a standard desktop computer (MacBook Pro M2, 16 GB RAM).
GenoPHI provides a unified command-line interface accessible through the genophi command:
# View available commands
genophi --help
# Get help for a specific command
genophi protein-family-workflow --helpFor most phage-host interaction prediction tasks, use these recommended settings:
genophi protein-family-workflow \
--input_path_strain strain_fastas/ \
--input_path_phage phage_fastas/ \
--phenotype_matrix interactions.csv \
--output_dir results/ \
--threads 8 \
--num_features 100 \
--num_runs_fs 25 \
--num_runs_modeling 50 \
--method rfe \
--use_clustering \
--cluster_method hierarchical \
--n_clusters 20 \
--filter_type strain \
--use_shapKey Parameters Explained:
--num_features 100: Select top 100 features (adjust based on dataset size)--num_runs_fs 25: 25 iterations for robust feature selection--num_runs_modeling 50: 50 modeling runs for reliable performance estimates--method rfe: Recursive Feature Elimination (balanced performance)--use_clustering: Enable sample clustering-aware filtering--filter_type strain: Critical for phage-host prediction - Ensures train/test splits separate by strain so the model learns to predict on new strains it hasn't seen before--use_shap: Generate SHAP plots and feature importance analysis for model interpretability
Note: For phage-host interaction prediction, --filter_type strain is strongly recommended. This controls how train/test splits are made during feature selection and modeling, ensuring the model never sees the same strain in both training and testing. This forces the model to learn generalizable patterns rather than memorizing specific strain characteristics.
For single-strain phenotypes (no phage data):
genophi protein-family-workflow \
--input_path_strain strain_fastas/ \
--phenotype_matrix phenotypes.csv \
--output_dir results/ \
--threads 8 \
--sample_column strain \
--phenotype_column resistance \
--filter_type noneGenoPHI provides the following main commands:
| Command | Description |
|---|---|
protein-family-workflow |
Recommended basic workflow: Complete protein family-based workflow |
full-workflow |
Protein families → k-mers from predictive proteins |
kmer-workflow |
Complete k-mer-based workflow from all proteins |
cluster |
Generate protein family clusters and feature tables |
select-features |
Perform feature selection on any feature table |
train |
Train predictive models on selected features |
predict |
Predict phenotypes using trained models |
select-and-train |
Feature selection + modeling from any feature table |
assign-features |
Assign features to new genomes |
assign-predict |
Assign features and predict (protein families) |
annotate |
Annotate predictive features with functional info |
kmer-assign-features |
Assign k-mer features to new genomes |
kmer-assign-predict |
Assign k-mer features and predict |
kmer-analysis |
Analyze k-mer composition and diversity |
The primary workflow for most applications. Performs complete protein family clustering, feature selection, and modeling.
genophi protein-family-workflow \
--input_path_strain strain_fastas/ \
--input_path_phage phage_fastas/ \
--phenotype_matrix interactions.csv \
--output_dir results/ \
--threads 8 \
--num_features 100 \
--num_runs_fs 25 \
--num_runs_modeling 50 \
--method rfe \
--filter_type strain \
--use_shapOutput Structure:
results/
├── strain/ # Strain MMseqs2 outputs
├── phage/ # Phage MMseqs2 outputs (if provided)
├── merged/ # Merged strain+phage feature table directory (if phage input)
│ └── full_feature_table.csv
├── feature_selection/ # Selected features and occurrence counts
│ ├── filtered_feature_tables/
│ └── features_occurrence.csv
├── modeling_results/ # Models and performance metrics
│ ├── cutoff_3/, cutoff_4/, cutoff_5/, ...
│ ├── model_performance/ # Summary plots, metrics, predictive_proteins/
│ ├── select_features_model_performance.csv
│ └── select_features_model_predictions.csv
├── workflow_report.txt # Runtime/performance summary
└── workflow_report.csv # Parameters and runtime metrics
For strain-level phenotypes (no phage data required):
genophi protein-family-workflow \
--input_path_strain strain_fastas/ \
--phenotype_matrix strain_phenotypes.csv \
--output_dir results/ \
--threads 8 \
--sample_column strain \
--phenotype_column antibiotic_resistance \
--task_type classification \
--filter_type nonePhenotype Matrix Format:
strain,antibiotic_resistance
Strain_001,1
Strain_002,0
Strain_003,1For regression:
--task_type regression \
--phenotype_column growth_rateThis workflow first identifies predictive protein families, then extracts k-mers specifically from those families for refined modeling. This combines the interpretability of protein families with the resolution of k-mer analysis.
genophi full-workflow \
--input_strain strain_fastas/ \
--input_phage phage_fastas/ \
--phenotype_matrix interactions.csv \
--output results/ \
--k 5 \
--threads 8Workflow Steps:
- Cluster proteins into families
- Perform feature selection on protein families
- Extract k-mers from predictive protein families only
- Train models on k-mer features
- Generate annotations for predictive k-mers
Generate k-mer features from all proteins without prior protein family analysis:
genophi kmer-workflow \
--input_strain_dir strain_fastas/ \
--input_phage_dir phage_fastas/ \
--phenotype_matrix interactions.csv \
--output kmer_results/ \
--k 4 \
--threads 8 \
--num_features 100 \
--num_runs_fs 25 \
--num_runs_modeling 50K-mer Specific Parameters:
--k 4: K-mer length (default: 4)--k_range: Generate k-mers from length 3 to k--one_gene: Include features with only one gene (default: False)
Advanced Options:
--use_dynamic_weights \ # Apply dynamic weighting
--weights_method inverse_frequency \ # Weighting method
--no-clustering \ # Disable clustering-aware train/test splitting
--use_shap # Save SHAP-based interpretation outputsgenophi cluster \
--input_strain strain_fastas/ \
--input_phage phage_fastas/ \
--phenotype_matrix interactions.csv \
--output clustering_results/ \
--min_seq_id 0.4 \
--coverage 0.8 \
--sensitivity 7.5 \
--threads 8Clustering Parameters:
--min_seq_id 0.4: Minimum sequence identity (range: 0-1)--coverage 0.8: Minimum coverage (range: 0-1)--sensitivity 7.5: MMseqs2 sensitivity (higher = more sensitive, slower)
Feature selection works on any feature table with a phenotype column:
genophi select-features \
--input feature_table.csv \
--output feature_selection/ \
--method rfe \
--num_features 100 \
--num_runs 25 \
--filter_type strain \
--phenotype_column interaction \
--threads 8Feature Selection Methods:
rfe: Recursive Feature Elimination (recommended)shap_rfe: RFE using SHAP valuesselect_k_best: ANOVA F-test (fast)chi_squared: Chi-squared testlasso: L1 regularizationshap: Direct SHAP importance
Advanced Selection Options:
--use_dynamic_weights \ # Handle imbalanced features
--weights_method inverse_frequency \ # Weighting strategy
--no-clustering \ # Disable clustering (enabled by default)
--cluster_method hierarchical \ # Clustering algorithm
--n_clusters 20 # Number of clustersTrain models from selected features (directory input):
genophi train \
--input feature_selection/filtered_feature_tables \
--output models/ \
--num_runs 50 \
--phenotype_column interaction \
--threads 8For regression tasks:
--task_type regression \
--phenotype_column efficiencyAdvanced Training Options:
--set_filter strain \ # Filter type: none, strain, phage (default: strain)
--use_dynamic_weights \ # Apply dynamic feature weighting
--weights_method inverse_frequency \ # Weighting: log10, inverse_frequency, balanced
--no-clustering \ # Disable clustering-aware splits
--cluster_method hierarchical \ # Clustering: hdbscan, hierarchical (default: hierarchical)
--n_clusters 20 # Number of clusters (default: 20)Run feature selection and modeling together from any feature table:
genophi select-and-train \
--full_feature_table custom_feature_table.csv \
--output results/ \
--method rfe \
--num_features 100 \
--num_runs_fs 25 \
--num_runs_modeling 50 \
--phenotype_column your_phenotype \
--sample_column your_sample_id \
--threads 8This command is flexible and works with:
- Protein family features
- K-mer features
- Custom features
- Any feature table with any phenotype / output column
Generate predictions for new genome combinations using pre-assigned features:
genophi predict \
--input_dir strain_feature_tables/ \
--model_dir models/cutoff_10 \
--output_dir predictions/ \
--phage_feature_table phage_features.csv \
--threads 8Parameters:
--input_dir: Directory with strain-specific feature tables--model_dir: Directory containing trained models--phage_feature_table: Path to phage feature table (optional for single-strain mode)--strain_source: Prefix for strain features (default: strain)--phage_source: Prefix for phage features (default: phage)
genophi assign-predict \
--input_dir new_strains/ \
--mmseqs_db results/tmp/strain/mmseqs_db \
--clusters_tsv results/strain/clusters.tsv \
--feature_map results/strain/features/selected_features.csv \
--tmp_dir tmp_assign/ \
--model_dir results/modeling_results/cutoff_10 \
--phage_feature_table results/phage/features/feature_table.csv \
--output_dir predictions/ \
--genome_type strainFor new phages:
--input_dir new_phages/ \
--mmseqs_db results/tmp/phage/mmseqs_db \
--clusters_tsv results/phage/clusters.tsv \
--tmp_dir tmp_assign_phage/ \
--strain_feature_table results/strain/features/feature_table.csv \
--output_dir predictions/ \
--genome_type phagegenophi kmer-assign-predict \
--input_dir new_strains/ \
--mmseqs_db results/tmp/strain/mmseqs_db \
--clusters_tsv results/strain/clusters.tsv \
--feature_map results/strain/features/selected_features.csv \
--filtered_kmers kmer_analysis/strain/filtered_kmers.csv \
--aa_sequence_file kmer_results/strain_combined.faa \
--tmp_dir tmp_kmer_assign/ \
--model_dir kmer_results/modeling/modeling_results/cutoff_10 \
--output_dir predictions/ \
--genome_type strainIdentifies proteins associated predictive protein families or k-mers and merges with functional information:
genophi annotate \
--feature_file_path results/feature_selection/filtered_feature_tables/select_feature_table_cutoff_10.csv \
--feature2cluster_path results/strain/features/selected_features.csv \
--cluster2protein_path results/strain/clusters.tsv \
--fasta_dir_or_file strain_fastas/ \
--modeling_dir results/modeling_results/cutoff_10 \
--annotation_table_path annotations.csv \
--output_dir annotations/ \
--feature_type strainProtein sequences in FASTA format (.faa files):
>protein_id_1
MKTAYIAKQRQISFVKSHFSRQLEERLGLIEVQAPILSRVGDGTQDNLSGAEKAVQVKVKALPDAQFEVVHSLAKWKRQ...
>protein_id_2
MRISTTITTTITITTGNGAG...
Important: Protein IDs must be unique across all genomes. If duplicates exist, GenoPHI will automatically prefix them with genome names.
Binary classification (infection/no infection):
strain,phage,interaction
Strain_001,Phage_A,1
Strain_001,Phage_B,0
Strain_002,Phage_A,1Regression (infection efficiency):
strain,phage,efficiency
Strain_001,Phage_A,0.85
Strain_001,Phage_B,0.12
Strain_002,Phage_A,0.93Classification:
strain,antibiotic_resistance
Strain_001,1
Strain_002,0
Strain_003,1Regression:
strain,growth_rate
Strain_001,0.42
Strain_002,0.38
Strain_003,0.51Column Names: Use --strain_column, --phage_column, --sample_column, and --phenotype_column to specify your column names.
| Method | Description | Best For | Speed |
|---|---|---|---|
| RFE (recommended) | Recursive Feature Elimination | General use, balanced performance | Medium |
| SHAP-RFE | RFE using SHAP values | Model-agnostic importance | Slow (High RAM) |
| SelectKBest | ANOVA F-test | Fast screening, linear relationships | Fast |
| Chi-squared | χ² test for independence | Categorical features | Fast |
| Lasso | L1 regularized regression | Sparse models, multicollinearity | Fast |
| SHAP | Shapley Additive Explanations | Direct feature importance | Slow (High RAM) |
Handle imbalanced feature distributions:
--use_dynamic_weights \
--weights_method inverse_frequency # or log10, balancedWhen to use:
- Features with highly variable occurrence frequencies
- Some features present in most genomes, others very rare
- Imbalanced positive/negative examples
Group correlated features for more robust selection:
--use_clustering \
--cluster_method hierarchical \ # or hdbscan
--n_clusters 20HDBSCAN Options:
--cluster_method hdbscan \
--min_cluster_size 5 \
--min_samples 5 \
--cluster_selection_epsilon 0.0-
AUC-ROC (Area Under ROC Curve): Overall discriminative ability
$$\text{AUC} = \int_{0}^{1} \text{TPR}(\text{FPR}) , d\text{FPR}$$ -
MCC (Matthews Correlation Coefficient): Balanced metric for all confusion matrix elements
$$\text{MCC} = \frac{TP \times TN - FP \times FN}{\sqrt{(TP + FP)(TP + FN)(TN + FP)(TN + FN)}}$$ -
Precision: Proportion of true positives among predicted positives
$$\text{Precision} = \frac{TP}{TP + FP}$$ -
Recall (Sensitivity): Proportion of true positives among actual positives
$$\text{Recall} = \frac{TP}{TP + FN}$$ -
F1 Score: Harmonic mean of precision and recall
$$F1 = 2 \times \frac{\text{Precision} \times \text{Recall}}{\text{Precision} + \text{Recall}}$$ -
Accuracy: Overall correct predictions
$$\text{Accuracy} = \frac{TP + TN}{TP + TN + FP + FN}$$
- RMSE: Root Mean Squared Error
- MAE: Mean Absolute Error
- R²: Coefficient of determination
GenoPHI generates comprehensive visualizations:
Per-Run Plots (modeling_results/cutoff_*/run_*/):
- Confusion matrices (classification)
- ROC curves with AUC scores
- Precision-Recall curves
- SHAP feature importance bar plots
- SHAP value scatter plots (beeswarm)
Summary Plots (modeling_results/model_performance/):
- SHAP beeswarm plots across all runs
- ROC curve comparisons across feature selection cutoffs
- Precision-Recall curve comparisons
- Hit rate and hit ratio curves
Example visualizations from the original README:
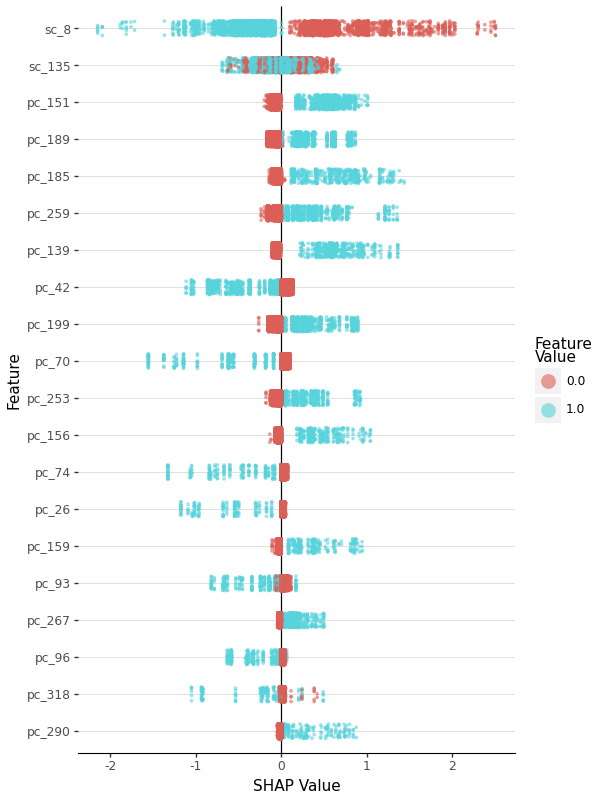 SHAP feature importance summary
SHAP feature importance summary
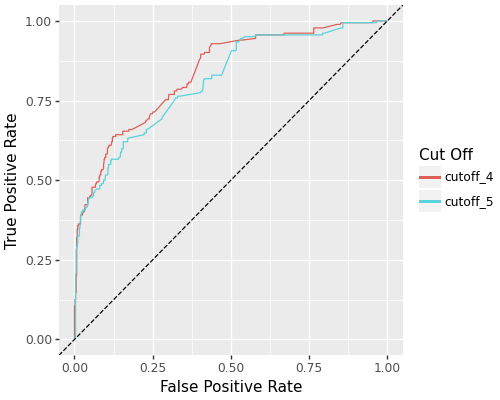
ROC curves comparing different feature selection cutoffs
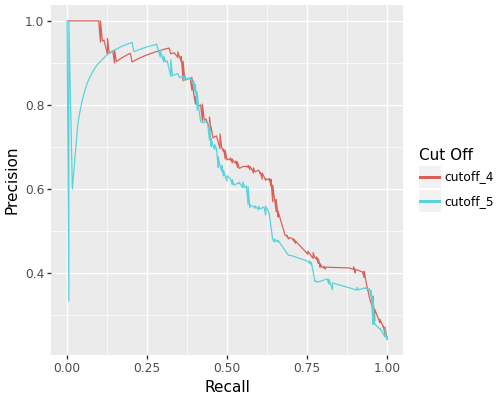 Precision-Recall curves
Precision-Recall curves
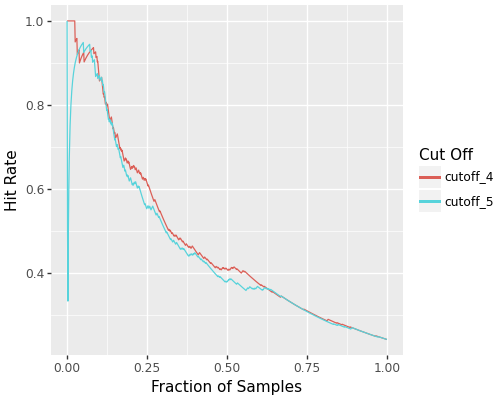 Hit rate analysis
Hit rate analysis
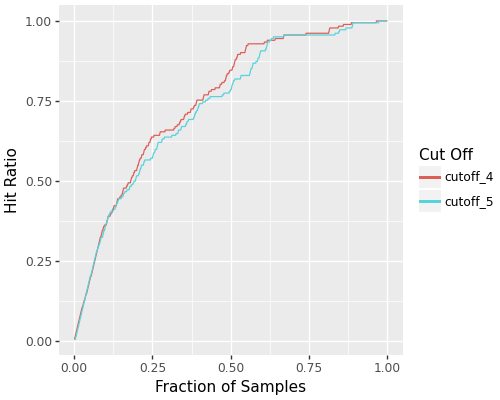 Hit ratio analysis
Hit ratio analysis
output_dir/
├── strain/
│ ├── clusters.tsv
│ ├── presence_absence_matrix.csv
│ └── features/
│ ├── feature_table.csv
│ ├── selected_features.csv
│ └── feature_assignments.csv
├── phage/ # Created when --input_path_phage is provided
│ ├── clusters.tsv
│ ├── presence_absence_matrix.csv
│ └── features/
│ ├── feature_table.csv
│ ├── selected_features.csv
│ └── feature_assignments.csv
├── merged/ # Created when phage input is provided
│ └── full_feature_table.csv
├── full_feature_table.csv # Created for single-strain mode (no phage input)
├── feature_selection/
│ ├── filtered_feature_tables/
│ │ ├── select_feature_table_cutoff_3.csv
│ │ ├── select_feature_table_cutoff_10.csv
│ │ └── ...
│ └── features_occurrence.csv
├── modeling_results/
│ ├── cutoff_3/, cutoff_4/, cutoff_5/, ...
│ ├── model_performance/
│ │ ├── model_performance_metrics.csv
│ │ └── predictive_proteins/
│ ├── select_features_model_performance.csv
│ └── select_features_model_predictions.csv
├── tmp/
│ ├── strain/
│ └── phage/ # Created when phage input provided
├── workflow_report.txt
└── workflow_report.csv
output_dir/
├── strain_combined.faa
├── strain_proteins.csv
├── phage_combined.faa # Optional
├── phage_proteins.csv # Optional
├── feature_tables/
│ ├── strain_feature_table.csv
│ ├── final_feature_table.csv
│ ├── phage_feature_table.csv # Optional
│ ├── phage_final_feature_table.csv # Optional
│ └── selected_features.csv
├── full_feature_table.csv
├── modeling/
│ ├── feature_selection/
│ └── modeling_results/
│ ├── cutoff_*/
│ ├── model_performance/model_performance_metrics.csv
│ ├── select_features_model_performance.csv
│ └── select_features_model_predictions.csv
├── workflow_report.txt
└── kmer_workflow_report.csv
GenoPHI can also be used programmatically:
from genophi.workflows import (
run_kmer_workflow,
run_modeling_workflow_from_feature_table,
assign_predict_workflow
)
from genophi.workflows.protein_family_workflow import run_protein_family_workflow
# Recommended: Protein family workflow
run_protein_family_workflow(
input_path_strain="strain_fastas/",
input_path_phage="phage_fastas/",
phenotype_matrix="interactions.csv",
output_dir="results/",
threads=8,
num_features=100,
num_runs_fs=50,
num_runs_modeling=100,
method='rfe',
filter_type='strain'
)
# _K_-mer workflow
run_kmer_workflow(
input_strain_dir="strain_fastas/",
input_phage_dir="phage_fastas/",
phenotype_matrix="interactions.csv",
output_dir="kmer_results/",
k=5,
threads=8,
num_features=100
)
# Feature selection and modeling from any feature table
run_modeling_workflow_from_feature_table(
full_feature_table="custom_features.csv",
output_dir="modeling_results/",
phenotype_column="your_phenotype",
sample_column="your_sample_id",
num_features=100,
num_runs_fs=50,
num_runs_modeling=100,
method='rfe'
)
# Prediction workflow
assign_predict_workflow(
input_dir="new_genomes/",
mmseqs_db="results/tmp/strain/mmseqs_db",
clusters_tsv="results/strain/clusters.tsv",
feature_map="results/strain/features/selected_features.csv",
tmp_dir="tmp_assign/",
model_dir="results/modeling_results/cutoff_10",
output_dir="predictions/",
genome_type='strain',
phage_feature_table_path="results/phage/features/feature_table.csv"
)Control how data is split during training and testing:
--filter_type none # Random split (default, but not recommended for phage-host)
--filter_type strain # Leave-strain-out CV splits - RECOMMENDED for phage-host
--filter_type phage # Leave-phage-out CV splitsImportant Recommendation: For phage-host interaction prediction, always use --filter_type strain. This controls how train/test splits are made during each iteration:
strain: Ensures the same strain never appears in both training and testing sets within an iteration. Forces the model to learn features that generalize to completely new bacterial strains.none: Random splits allow the same strains in both sets, leading to overly optimistic performance because the model can memorize strain-specific patterns.phage: Leave-phage-out splits, useful for testing generalization to new phages.
The split type fundamentally changes what the model learns, not just how it's evaluated.
Models use grid search for hyperparameter optimization. Default parameters are optimized for phage-host interaction prediction but can be customized in the code.
For large datasets, adjust memory and threading:
--max_ram 64 \ # Maximum RAM in GB
--threads 16 \ # Number of CPU threads
--clear_tmp # Remove temporary files after completionFor continuous phenotypes:
--task_type regression \
--phenotype_column efficiencyGenoPHI will use appropriate regression metrics (RMSE, MAE, R²) instead of classification metrics.
Filter features by cluster presence before modeling:
--use_feature_clustering \
--feature_cluster_method hierarchical \
--feature_n_clusters 20 \
--feature_min_cluster_presence 2This removes features that appear in fewer than feature_min_cluster_presence genome clusters.
This option set is available in protein-family and full workflows (protein-family-workflow, cluster, and full-workflow).
To help you get started, we recommend testing GenoPHI with:
Minimal test case with 10 strains, 5 phages:
test_data/
├── strains/
│ ├── Strain_001.faa
│ ├── Strain_002.faa
│ └── ...
├── phages/
│ ├── Phage_A.faa
│ ├── Phage_B.faa
│ └── ...
└── interactions.csv
Issue: MMseqs2 not found
Solution: Ensure MMseqs2 is installed and in your PATH
conda install -c bioconda mmseqs2
which mmseqs # Should show the path
Issue: Out of memory errors
Solution:
- Reduce --max_ram parameter
- Process fewer genomes at once
- Use --clear_tmp to remove intermediate files
- Increase system swap space
Issue: Duplicate protein IDs
Solution: GenoPHI automatically detects and prefixes duplicates with genome names
If you want to prevent this, ensure protein IDs are unique across all input files
Issue: No predictive features found
Solution:
- Try different feature selection methods (--method)
- Adjust num_features parameter
- Check that phenotype matrix has sufficient positive/negative examples
- Verify that interaction matrix matches genome filenames
Issue: Poor model performance
Solution:
- Increase num_runs_fs and num_runs_modeling for more robust results
- Try different feature selection methods
- Use --use_dynamic_weights for imbalanced features
- Enable --use_clustering for feature grouping
- Check data quality and ensure phenotype matrix is correct
- Try different clustering parameters (min_seq_id, coverage)
Issue: Models take too long to train
Solution:
- Reduce num_runs_modeling
- Reduce num_features
- Increase --threads parameter
- Use faster feature selection methods (select_k_best, chi_squared)
GenoPHI includes a comprehensive test suite organized into multiple tiers for different testing scenarios.
# Verify installation
pytest -m smoke -v
# Run all tests (requires MMSeqs2)
pytest -v- Smoke tests (<5 seconds): Package installation verification
- Integration tests (~30-45 min): Module-to-module interactions
- End-to-end tests (~60-90 min): Complete workflow validation
See tests/README.md for detailed testing documentation, including:
- How to run specific test tiers
- Test data organization
- Baseline metrics for regression testing
- CI/CD recommendations
pytest -m smoke # Quick installation check
pytest -m integration # Module integration tests
pytest -m e2e # End-to-end workflows
pytest -m "not requires_mmseqs2" # Skip MMSeqs2-dependent testsQ: Can GenoPHI be used for organisms other than phages and bacteria?
A: Yes! While designed for phage-host interactions, GenoPHI works with any protein sequences and phenotypes.
Q: How many genomes do I need for reliable predictions?
A: Minimum: ~20 strains and 20 phages with ~400 interactions. Recommended: 50+ strains, 50+ phages, 5000+ interactions for robust models.
Q: What file formats are required?
A: FASTA files (.faa) for protein sequences and CSV for phenotype matrices. See Input Data Formats for details.
Q: What's the difference between protein family and k-mer approaches?
A: Protein families group similar full-length proteins (interpretable, captures protein-level patterns). K-mers analyze short amino acid sequences (high resolution, captures local patterns). The full-workflow combines both by extracting k-mers from predictive protein families.
Q: Should I use single-strain or phage-host mode?
A: Use phage-host mode for interaction prediction. Use single-strain mode for strain-level phenotypes (resistance, growth rate, etc.) where phage data isn't relevant.
Q: Which feature selection method should I use?
A: Start with RFE (balanced performance).
Q: How do I interpret SHAP plots?
A: SHAP beeswarm plots show feature importance. Features at the top are most important. Red dots = high feature values, blue = low. Position right of center = positive impact on prediction. Enable with --use_shap flag during model training.
Q: Can I use custom features instead of protein families?
A: Yes! Use select-and-train with any feature table containing a phenotype column (metabolic pathways, gene presence/absence, etc.).
Q: How do I handle imbalanced datasets?
A: Use --use_dynamic_weights with --weights_method inverse_frequency to balance feature importance. CatBoost also has built-in class balancing.
Q: Models perform poorly - what should I try?
A: (1) Increase num_runs for more robust estimates, (2) Try different clustering parameters, (3) Enable dynamic weighting, (4) Check data quality and phenotype matrix accuracy.
Q: How much RAM do I need?
A: Minimum 8 GB. Recommend 16+ GB for 50+ genomes, 32+ GB for 100+ genomes. Use --max_ram to limit memory usage.
- Start with recommended defaults for initial analysis
- For phage-host predictions: Always use
--filter_type strain- This forces the model to learn generalizable patterns by ensuring strains in the test set are never seen during training (critical!) - Run multiple iterations (
num_runs_fs = 25,num_runs_modeling = 50) for robust results - Enable clustering (
--use_clustering) for correlated features - Check data quality before modeling - ensure phenotype matrix matches genome filenames exactly
- Use SHAP plots (
--use_shap) to understand which features drive predictions and for model interpretability - For single-strain phenotypes: Use
--filter_type noneor omit (random splits are appropriate when no phage data)
- First stable release
- Protein family-based workflow with MMseqs2 clustering
- K-mer-based workflow with flexible k-mer lengths
- Multiple feature selection methods (RFE, SHAP, SelectKBest, Chi-squared, Lasso)
- CatBoost model training with hyperparameter optimization
- SHAP-based interpretability
- Support for classification and regression tasks
- Single-strain and phage-host prediction modes
- Unified CLI with 14 commands
- Comprehensive visualization outputs
- Web interface for prediction and visualization
- Docker container for easy deployment
The datasets used in the GenoPHI publication are included in the data/ directory for reproducibility and benchmarking purposes.
data/
├── experimental_validation/
│ ├── BASEL_ECOR_interaction_matrix.csv # BASEL collection against ECOR strains for model validation
│ └── ECOR27_TnSeq_high_fitness_genes.csv # Filtered RB-TnSeq results
├── interaction_matrices/
│ ├── ecoli_interaction_matrix.csv # E. coli phage-host interactions
│ ├── ecoli_interaction_matrix_subset.csv # Smaller E. coli subset for testing
│ ├── klebsiella1_interaction_matrix.csv # Klebsiella dataset 1
│ ├── klebsiella2_interaction_matrix.csv # Klebsiella dataset 2
│ ├── pseudomonas_interaction_matrix.csv # Pseudomonas interactions
│ └── vibrio_interaction_matrix.csv # Vibrionaceae interactions
└── test_data/ # Test datasets for test suite
Analysis scripts used to generate figures and results for the GenoPHI publication are available in the manuscript_scripts/ directory. These scripts demonstrate advanced usage patterns and reproduce the analyses presented in the paper.
If you use GenoPHI in your research, please cite:
@article{noonan2025genophi,
author = {Noonan, Avery J. C. and Moriniere, Lucas and Rivera-López, Edwin O. and Patel, Krish and Pena, Melina and Svab, Madeline and Kazakov, Alexey and Deutschbauer, Adam and Dudley, Edward G. and Mutalik, Vivek K. and Arkin, Adam P.},
title = {Phylogeny-agnostic strain-level prediction of phage-host interactions from genomes},
year = {2025},
doi = {10.1101/2025.11.15.688630},
publisher = {Cold Spring Harbor Laboratory},
url = {https://www.biorxiv.org/content/10.1101/2025.11.15.688630v1},
journal = {bioRxiv}
}Preprint: Noonan, A.J.C., Moriniere, L., Rivera-López, E.O., Patel, K., Pena, M., Svab, M., Kazakov, A., Deutschbauer, A., Dudley, E.G., Mutalik, V.K., & Arkin, A.P. (2025). Phylogeny-agnostic strain-level prediction of phage-host interactions from genomes. bioRxiv. https://doi.org/10.1101/2025.11.15.688630
We welcome contributions to GenoPHI! Here's how you can help:
- Report bugs or suggest features via GitHub Issues
- Submit pull requests for bug fixes or new features
- Improve documentation by fixing typos or adding examples
- Share your use cases and publications using GenoPHI
- Test on different platforms and report compatibility issues
- Fork the repository and create a feature branch
- Follow PEP 8 style guidelines for Python code
- Add tests for new functionality
- Update documentation as needed
- Submit a pull request with a clear description of changes
We are committed to providing a welcoming and inclusive environment. Please be respectful and constructive in all interactions.
This software is available under the MIT License. See the LICENSE file for details.
This software is subject to Lawrence Berkeley National Laboratory copyright. The U.S. Government retains certain rights as this software was developed under funding from the U.S. Department of Energy.
For questions, issues, or feature requests:
- Open an issue on GitHub
- Contact: Avery Noonan (averynoonan@gmail.com)
This was completed as part of the BRaVE Phage Foundry at Lawrence Berkeley National Laboratory which is supported by the U.S. Department of Energy, Office of Science, Office of Biological & Environmental Research under contract number DE-AC02-05CH11231. This work was also supported by the National Science Foundation (NSF) of the United States under grant award No. 2220735 (EDGE CMT: Predicting bacteriophage susceptibility from Escherichia coli genotype).